Wie Moleküle auf Licht reagieren – ein theoretischer Blick auf Photoschalter 💡
Motivation¶
Molekulare Photoschalter sind chemische Verbindungen, die auf Licht reagieren, indem sie ihre räumliche Struktur verändern. Dadurch ändern sich auch ihre chemischen und physikalischen Eigenschaften. Man kann sie sich wie winzige Kippschalter vorstellen – sie werden jedoch nicht mechanisch betätigt, sondern durch gezielte Lichtbestrahlung zwischen zwei Zuständen umgeschaltet.
Solche Systeme sind nicht nur faszinierend, sondern auch technologisch relevant. Sie kommen zum Beispiel in lichtaktivierbaren Arzneistoffen oder in Materialien zum Einsatz, die ihre Eigenschaften unter Bestrahlung gezielt anpassen.
In diesem Computerexperiment untersuchen Sie exemplarisch einige Photoschalter und berechnen deren Absorptionsspektren im sichtbaren und ultravioletten Bereich des elektromagnetischen Spektrums. So erhalten Sie einen ersten Einblick, wie Methoden der theoretischen Chemie zur strukturellen und spektroskopischen Analyse lichtinduzierter Schaltprozesse eingesetzt werden können.
Grundlagen¶
Azobenzol¶
In diesem Versuch arbeiten Sie mit dem Molekül Azobenzol. Es besteht aus zwei Phenylgruppen, die über eine Azobrücke (–N=N–) miteinander verbunden sind.

Azobenzol, dargestellt als Skelettformel.
Summen-, Struktur- und Skelettformeln
Azobenzol besteht aus 12 Kohlenstoff-, 10 Wasserstoff- und 2 Stickstoffatomen und besitzt somit die Summenformel C₁₂H₁₀N₂.
In den beiden Phenylringen ist jedes Kohlenstoffatom mit zwei weiteren Kohlenstoffatomen verbunden. Je ein Kohlenstoffatom pro Ring bindet die Azobrücke. Die übrigen Kohlenstoffatome tragen jeweils ein Wasserstoffatom.
Die genauen Bindungsverhältnisse lassen sich mit einer Strukturformel darstellen:

Die Strukturformel des Azobenzols.
Organische Moleküle besitzen häufig ein Grundgerüst aus Kohlenstoffatomen. Zur besseren Übersicht – und um Zeit beim Zeichnen zu sparen – lässt man in der Skelettformel die Kohlenstoffatome sowie die daran gebundenen Wasserstoffatome weg.
Azobenzol selbst ist farblos, bildet aber das Grundgerüst für zahlreiche Farbstoffe. Diese entstehen durch Substitution, also durch Austausch von Wasserstoffatomen gegen andere Atome oder Atomgruppen. Wird beispielsweise am vierten Kohlenstoffatom eines Phenylrings ein Wasserstoffatom durch eine Methoxygruppe (–OCH₃) ersetzt, entsteht eine gelbe Verbindung.

4-Methoxyazobenzol – ein gelber Farbstoff.
Nomenklatur
Jede chemische Verbindung kann eindeutig benannt werden; die entsprechenden Regeln werden von der IUPAC (International Union of Pure and Applied Chemistry) festgelegt.
Ein zentrales Element ist die Durchnummerierung der Kohlenstoffatome im Grundgerüst. Beim Azobenzol werden die Kohlenstoffatome des ersten Phenylrings mit 1–6 und die des zweiten Rings mit 1′–6′ nummeriert. Die Atome 1 und 1′ sind dabei immer die beiden Kohlenstoffatome, die direkt an die Azobrücke gebunden sind. In welcher Richtung anschließend weitergezählt wird (im oder gegen den Uhrzeigersinn) hängt normalerweise von den vorhandenen Substituenten ab – in diesem Versuch wird zur Vereinfachung jedoch eine feste Nummerierung verwendet.

Die in diesem Versuch verwendete Nummerierung der Kohlenstoffatome.
Wird nun beispielsweise am 2. Kohlenstoffatom ein Wasserstoff durch ein Chloratom ersetzt, erhält man die Verbindung 2-Chlorazobenzol.
In diesem Versuch treten die folgenden Substituenten auf:
| Summenformel | Abkürzung | Name des Substituenten |
|---|---|---|
| CH₃ | Me | Methyl |
| N(CH₃)₂ | NMe₂ | (Dimethylamino) |
| CF₃ | CF₃ | (Trifluormethyl) |
| OCH₃ | OMe | Methoxy |
| F | F | Fluor |
| SO₂CF₃ | SO₂CF₃ | (Trifluormethansulfonyl) |
Tritt ein Substituent mehrfach auf, wird dies durch Präfixe wie di-, tri- oder tetra- angezeigt. Wird z. B. beim 2-Chlorazobenzol zusätzlich am dritten Kohlenstoffatom des zweiten Rings ein weiteres Chloratom eingeführt, entsteht die Verbindung 2,3′-Dichlorazobenzol.
Mit diesen Regeln können Sie alle im Versuch vorkommenden Moleküle eindeutig benennen. Überprüfen Sie zur Übung, ob es sich bei der folgenden Verbindung um 3,5-Dimethyl-4-methoxy-2′-fluor-4′-(trifluormethyl)-5′-(dimethylamino)-azobenzol handelt:

Durch die Azobrücke kann Azobenzol in zwei unterschiedlichen räumlichen Anordnungen (Konfigurationen) vorliegen. Alle bisher gezeigten Strukturen entsprechen der trans-Form, bei der die beiden Phenylringe auf gegenüberliegenden Seiten der Azobrücke stehen. Bei der cis-Form befinden sich beide Ringsysteme auf derselben Seite.
E,Z-Nomenklatur
Eine andere, systematischere Bezeichnungsweise für die Anordnung von Substituenten an Doppelbindungen verwendet die Symbole E (für entgegen) und Z (für zusammen). Für unsere Zwecke reichen jedoch trans und cis aus.
Moleküle mit gleicher Summenformel und Molekülmasse, die sich jedoch in der räumlichen Anordnung oder Verknüpfung der Atome unterscheiden, bezeichnet man als Isomere.

Cis-Azobenzol.
Die trans-Form ist energetisch stabiler und wird unter normalen Bedingungen bevorzugt. Durch Bestrahlung mit Licht geeigneter Wellenlänge kann jedoch eine Umwandlung in die cis-Form ausgelöst werden. Diese lichtinduzierte Isomerisierung bildet die Grundlage für die photoschaltbaren Eigenschaften des Moleküls.
Ein Beispiel hierfür ist das oben erwähnte 4-Methoxyazobenzol: Bei Bestrahlung mit ultraviolettem Licht wechselt es von seiner gelblichen trans-Form in eine rot-braune cis-Form.

Die lichtinduzierte Isomerisierung des Azobenzols.
Dreidimensionale Visualisierung
Chemische Strukturformeln eignen sich gut zur Darstellung grundlegender räumlicher Eigenschaften wie der cis-trans-Isomerie, sind jedoch durch ihre zweidimensionale Darstellung begrenzt. Eine Alternative bietet die 3D-Visualisierung. So genannte Kalotten- oder Kugel-Stab-Modelle zeigen Moleküle im Raum. Durch Rotation und Bewegung des Moleküls – ähnlich wie in einem Computerspiel – entsteht ein umfassenderes räumliches Bild.
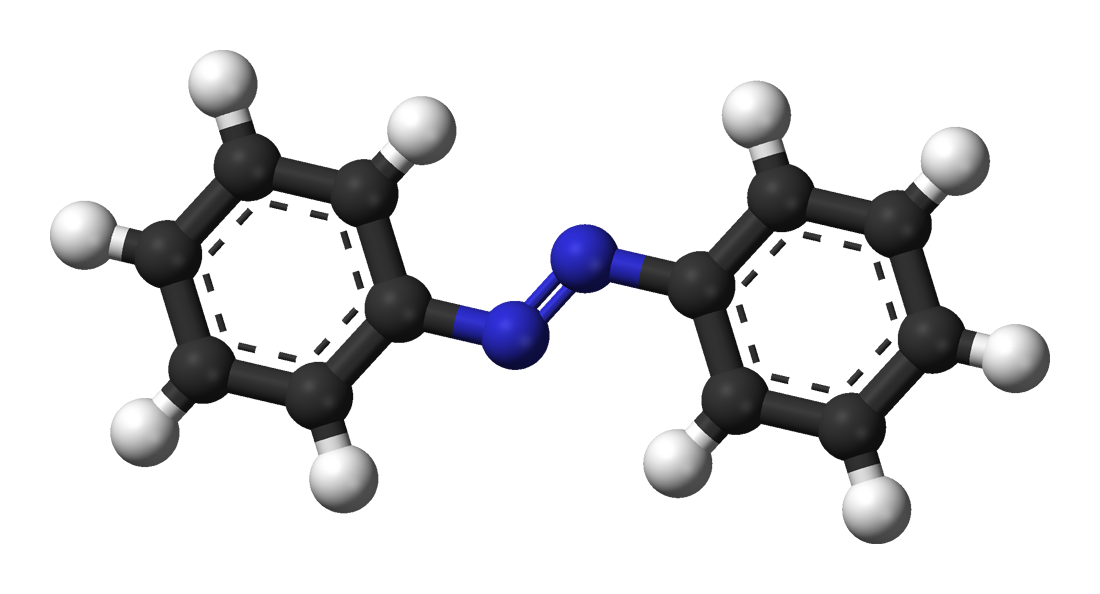
Kugel-Stab-Modell von trans-Azobenzol. Benjah-bmm27, Public Domain. Quelle: Wikimedia Commons.
Das elektromagnetische Spektrum¶
Das Licht, das wir mit dem Auge wahrnehmen können, ist nur ein kleiner Teil des sogenannten elektromagnetischen Spektrums. Dieses umfasst alle Formen elektromagnetischer Strahlung – von energiearmen Radiowellen über Mikrowellen und Infrarotstrahlung bis hin zu sichtbarem Licht, UV-Strahlung, Röntgenstrahlen und hochenergetischen Gammastrahlen.
Die einzelnen Bereiche unterscheiden sich in ihrer Energie bzw. Wellenlänge . Grundsätzlich gilt: je höher die Energie, desto kürzer die Wellenlänge. Dies wird durch folgende Gleichung beschrieben:
wobei die Planck-Konstante und die Lichtgeschwindigkeit im Vakuum ist. Das Produkt der beiden Konstanten beträgt
In der Atom- und Molekülphysik verwendet man häufig die Einheiten Elektronenvolt (eV) und Nanometer (nm), da sich damit deutlich handlichere Zahlenwerte ergeben als mit Joule bzw. Meter.

Das elektromagnetische Spektrum. Die obere Reihe zeigt (von links nach rechts) stilisierte Darstellungen von Gammastrahlen, Röntgenstrahlen, UV-Strahlung, sichtbarem Licht, Infrarotstrahlung, Mikrowellen und Radiowellen. Die untere Reihe vergrößert den sichtbaren Teil des Spektrums. Tatoute und Phrood~commonswiki, Lizenz: CC BY-SA 3.0. Quelle: Wikimedia Commons.
Absorption elektromagnetischer Strahlung¶
Moleküle können mit Licht wechselwirken, indem sie dieses absorbieren. Wie diese Wechselwirkung abläuft, wird durch die Gesetze der Quantenmechanik beschrieben. Eine der grundlegenden Erkenntnisse dieser Theorie ist, dass Moleküle nicht Licht beliebiger Energie absorbieren können, sondern nur ganz bestimmte Energien. Stimmt die Energie, so wird das Licht aufgenommen und das Molekül in einen um diesen Energiebetrag höheren energetischen Zustand versetzt. Schematisch lässt sich dieser Prozess wie folgt darstellen:
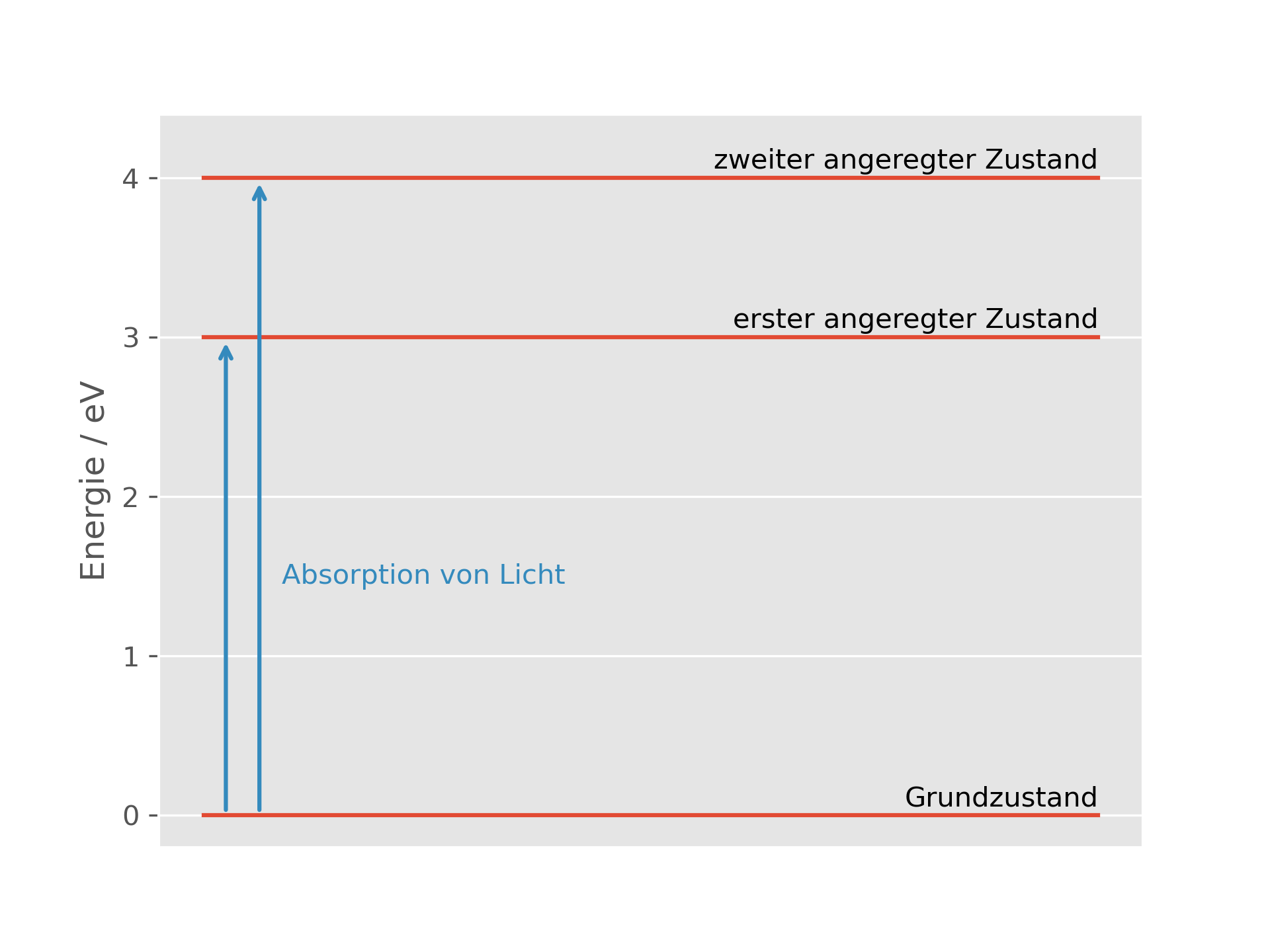
Jablonskitermschema eines Absorptionsprozesses. Das (fiktive) Molekül kann entweder Licht mit 3.0 eV oder 4.0 eV absorbieren, um in den ersten bzw. zweiten angeregten Zustand versetzt zu werden.
Trägt man auf, wie stark ein Molekül Licht mit einer bestimmten Energie absorbiert, so erhält man ein Linienspektrum.
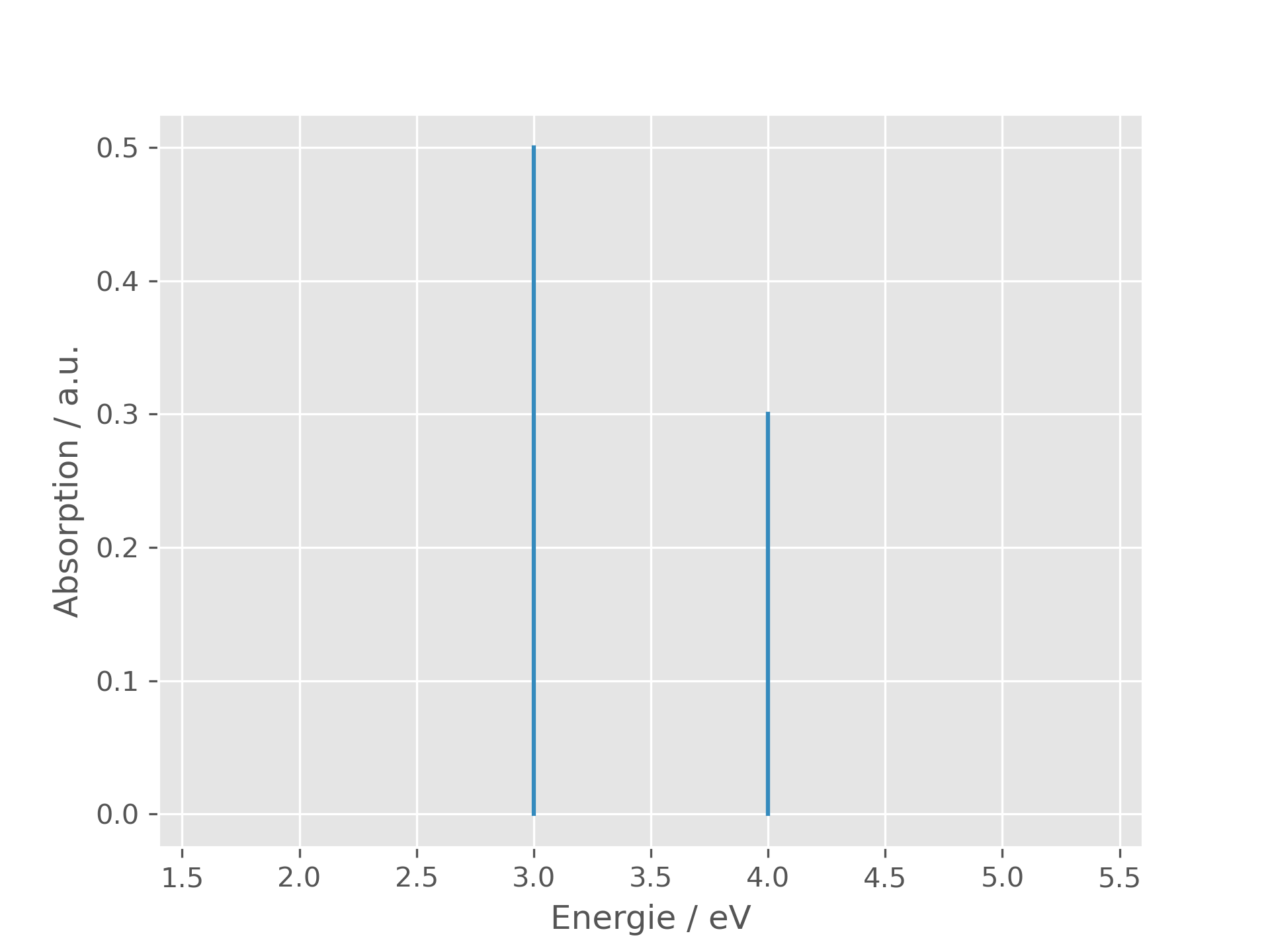
Das Spektrum eines Moleküls, das Licht mit 3.0 eV und 4.0 eV absorbiert. Die genauen Zahlenwerte der Absorption sind für unseren Versuch nicht relevant; daher wird die Einheit a.u. (arbitrary units) verwendet.
In experimentellen Absorptionsspektren erscheinen jedoch keine scharfen Linien, sondern breite Banden. Das liegt unter anderem daran, dass im Experiment viele miteinander wechselwirkende Moleküle untersucht werden – häufig sogar in Lösung. Außerdem bewegen sich Moleküle aufgrund der Temperatur ständig, wodurch die bevorzugte Übergangsenergie leicht variiert. Hinzu kommen instrumentelle Effekte (z. B. begrenzte Auflösung) sowie fundamentale quantenmechanische Effekte (z. B. endliche Lebensdauer der angeregten Zustände). All dies führt zu einer sogenannten Linienverbreiterung.
Es entsteht ein Bandenspektrum. Aus der Lage und Form dieser Banden lassen sich wichtige Informationen über den elektronischen Aufbau eines Moleküls gewinnen.
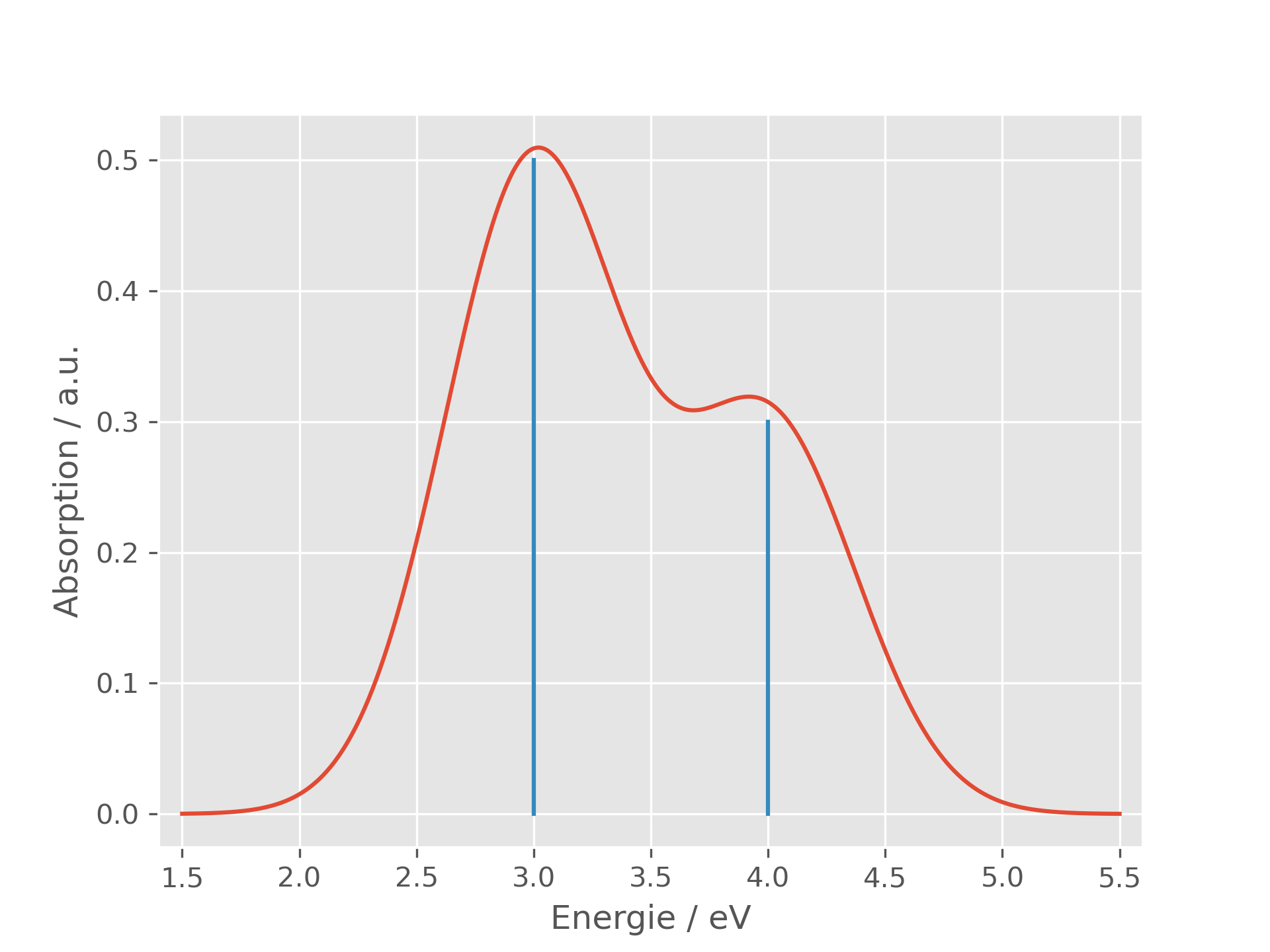
Das Bandenspektrum eines Moleküls, das bevorzugt Licht mit 3.0 eV und 4.0 eV absorbiert. Zur besseren Orientierung wurde das zugrunde liegende Linienspektrum im Hintergrund dargestellt.
Für besonders Interessierte: Absorption, Transmission, Extinktion, Übergangswahrscheinlichkeiten
Absorption bezeichnet die Aufnahme von Licht durch ein Molekül. Transmission ist der Anteil des Lichts, der vom Molekül nicht aufgenommen wird und hindurchtritt. Die Extinktion beschreibt die gesamte Abschwächung der Strahlung und umfasst sowohl Absorption als auch Streuung. In spektroskopischen Experimenten wird häufig die Extinktion genutzt, da sie direkt von der Konzentration der absorbierenden Substanz abhängt (Lambert-Beer-Gesetz, siehe auch PC-Versuch).
In diesem Computerversuch wird die Übergangswahrscheinlichkeit berechnet, also die Wahrscheinlichkeit, dass ein Molekül durch Licht in einen höheren energetischen Zustand angeregt wird. Diese Größe ist direkt proportional zur Absorption.
Motivation: Das elektromagnetische Fenster der Atmosphäre
Absorptionsspektren spielen nicht nur bei der Charakterisierung einzelner Moleküle eine Rolle, sondern sind auch entscheidend für das Verständnis des Energiehaushalts unserer Erde – und damit des Klimawandels.
Eines der wichtigsten Beispiele ist das Absorptionsspektrum unserer Atmosphäre. Die folgende Darstellung zeigt die relative Durchlässigkeit (Transmission) für elektromagnetische Strahlung. Das gelb markierte elektromagnetische Fenster ist dabei von besonderer Bedeutung für den Wärmehaushalt unseres Planeten.

Atmosphärische Durchlässigkeit. PNG-Version: Herbertweidner; SVG-Umsetzung: Cepheiden. Quelle/Lizenz: Wikimedia Commons.
Das elektromagnetische Fenster ist deshalb so wichtig, weil nur in diesem Bereich die Sonnenstrahlung nahezu ungehindert zur Erdoberfläche gelangen kann – und gleichzeitig die von der Erde abgegebene Wärmestrahlung wieder ins Weltall entweichen kann. Würde die Atmosphäre in diesem Bereich stärker absorbieren, würde sich die Erde deutlich stärker aufheizen. 🌍
Theoretische Chemie¶
Die theoretische Chemie beschäftigt sich mit chemischen Fragestellungen, die mithilfe nichtexperimenteller Methoden
untersucht werden.
Dazu gehören:
grundlegende Theorien wie die Quantenmechanik oder statistische Mechanik,
computergestützte Berechnungen und Simulationen, mit denen Molekülstrukturen, Energien oder Reaktivitäten bestimmt werden,
die Modellierung komplexer Systeme (z. B. Protein-Docking oder Drug-Design)
sowie vereinfachte, qualitative Modelle, mit denen sich allgemeine Trends abschätzen lassen.
In diesem Praktikumsversuch werden Sie verschiedene solcher Berechnungen durchführen.
Wie die zugrundeliegenden Methoden im Detail funktionieren, lernen Sie im Verlauf Ihrer theoretisch-chemischen
Ausbildung (3./4. Semester, optional 5./6. Semester sowie im Masterstudium).
Für diesen Versuch können Sie sich die eingesetzten Verfahren zunächst als Blackbox vorstellen:
Der innere Aufbau muss nicht bekannt sein – wichtig ist lediglich, dass bestimmte Eingaben (z. B. eine Molekülstruktur)
zu bestimmten Ausgaben führen (z. B. Energie oder Spektrum).

Krauss, Lizenz: CC BY-SA 4.0. Quelle: Wikimedia Commons.
Atomkoordinaten¶
Strukturformeln sind für Chemiker*innen zwar intuitiv verständlich, für die maschinelle Weiterverarbeitung jedoch ungeeignet. Deutlich besser geeignet sind atomare Koordinaten, d. h. die Positionen (x, y, z) eines jeden Atoms im Raum.
Das Strukturerstellungswerkzeug erzeugt für Sie solche atomaren Koordinaten im sogenannten XYZ-Format. Dieses einfache Textformat ist wie folgt aufgebaut:
[Anzahl Atome]
[Kommentar oder Leerzeile]
[Elementsymbol des ersten Atoms] [X] [Y] [Z]
[Elementsymbol des zweiten Atoms] [X] [Y] [Z]
[...]Die Koordinaten werden dabei in der Einheit Ångström angegeben (). Die Struktur eines einfachen Wassermoleküls (H₂O) kann zum Beispiel wie folgt im XYZ-Format beschrieben werden:
3
O 0.000 0.000 0.000
H 0.000 -0.757 0.587
H 0.000 0.757 0.587Dieses Format zeigt, dass sich das Sauerstoffatom im Ursprung des Koordinatensystems befindet und dass die beiden Wasserstoffatome in der XY-Ebene liegen .
Das XYZ-Format dient in diesem Versuch zur Übergabe molekularer Strukturen zwischen den einzelnen Werkzeugen. Es fungiert damit sowohl als Eingabe- als auch als Ausgabeformat der jeweiligen Black Boxes. Sie selbst müssen keine XYZ-Dateien erstellen – sollten den grundlegenden Aufbau dieses Formats jedoch kennen und beschreiben können.
Molekulare Energien¶
Die vermutlich wichtigste Größe in der theoretischen Chemie ist die Energie eines Moleküls. Alle Moleküle bestehen aus Atomen – diese wiederum aus Atomkernen und Elektronen. Diese Teilchen wechselwirken miteinander: anziehende Wechselwirkungen senken die Energie des Systems, abstoßende Wechselwirkungen erhöhen sie. Ist die Summe aller Wechselwirkungen anziehend, so bleibt das Molekül stabil und zerfällt nicht in seine Einzelteile. Das ist bei allen hier betrachteten Molekülen der Fall – aus diesem Grund sind die berechneten Energien negativ.
Vom Betrag her erscheinen die berechneten Energien sehr groß (typischerweise in der Größenordnung
von ).
Das liegt daran, dass Energien immer relativ zu einem Bezugspunkt angegeben werden.
In theoretisch-chemischen Rechnungen ist dieser Bezugspunkt oft ein hypothetischer Zustand, bei dem alle Atomkerne und
Elektronen unendlich weit voneinander entfernt sind.
Bringt man diese Teilchen in Molekülform zusammen, wird eine sehr große Energiemenge frei – daher der große negative
Wert.
In der Realität betrachtet man jedoch häufig Prozesse, bei denen Moleküle ihre Konfiguration ändern oder durch chemische
Reaktionen ineinander übergehen.
Bei solchen Veränderungen spielen meist deutlich kleinere Energiemengen eine Rolle.
Die Energieunterschiede zwischen zwei Konfigurationen lassen sich einfach berechnen.
Liegt ein Azobenzolderivat beispielsweise in der cis-Konfiguration mit einer Energie von
vor und wechselt anschließend in die trans-Konfiguration mit
, so ergibt sich zunächst eine Energiedifferenz in eV:
Für das Protokoll berichten Sie solche Energiedifferenzen zwischen Konfigurationen in kJ/mol:
Ein negativer Wert bedeutet, dass die trans-Konfiguration energetisch günstiger ist (niedrigere Energie) – der Übergang von cis nach trans setzt also Energie frei.
Strukturoptimierung¶
Moleküle können viele verschiedene räumliche Strukturen annehmen, die sich in ihren Atompositionen – und damit auch in ihrer Energie – unterscheiden. Einige dieser Strukturen entsprechen lokalen Energieminima; ihre Energie lässt sich durch kleine Auslenkungen der Atomkoordinaten nicht weiter verringern. Im Falle des Azobenzols sind dies die Strukturen, die den cis- und trans-Konfigurationen zugeordnet werden können. Eine zentrale Aufgabe der computergestützten Chemie besteht darin, solche Minimumsstrukturen zu finden und zu charakterisieren – dieser Vorgang wird als Strukturoptimierung bezeichnet.
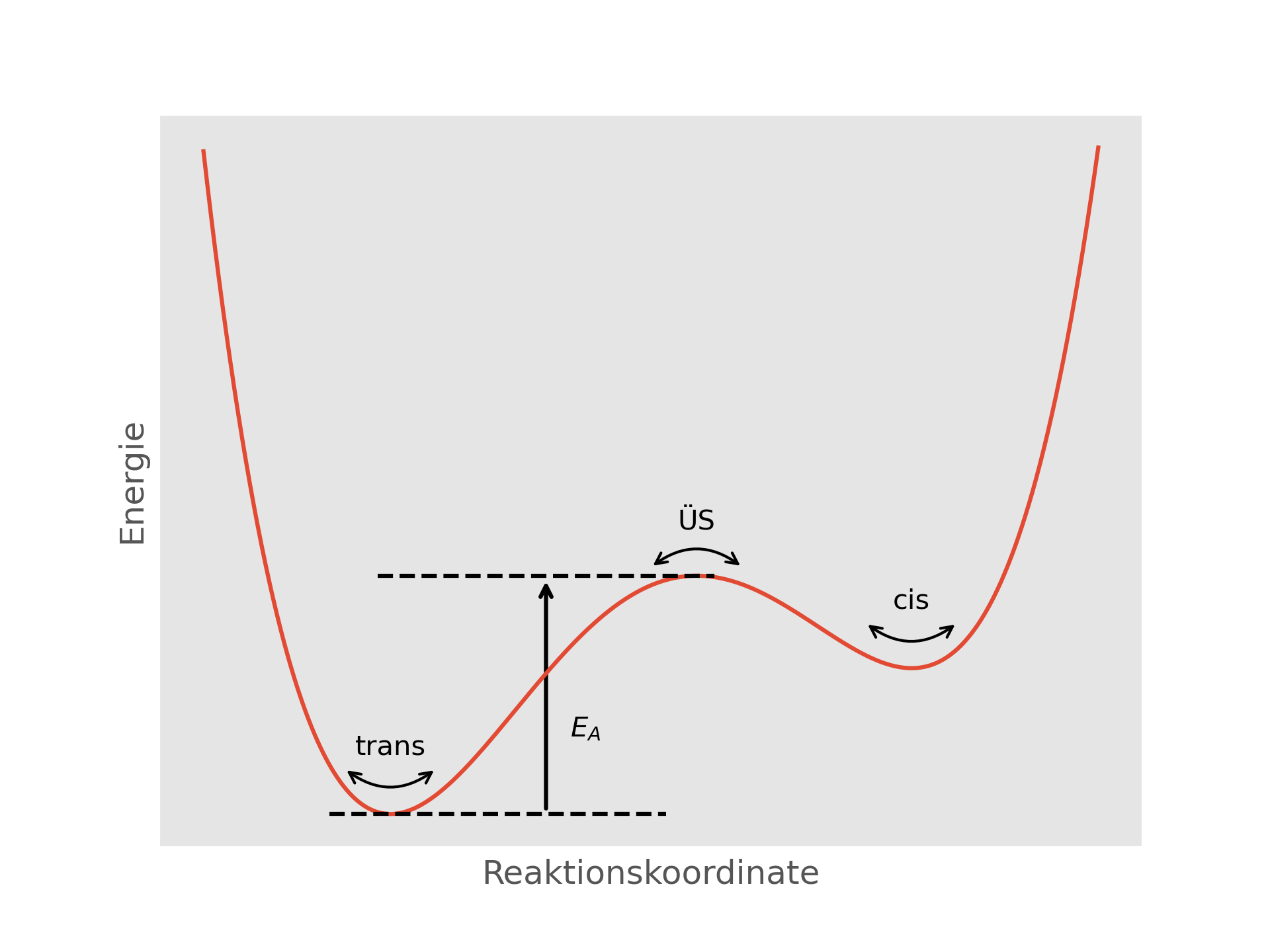
Könnte man die Auslenkung aller Atomkoordinaten auf eine einzelne Koordinate projizieren (Reaktionskoordinate), so würden die cis- und trans-Konfigurationen des Azobenzols den lokalen Minima dieser Funktion entsprechen. Schon kleinste geometrische Änderungen weg von diesen Strukturen führen zu einem Energieanstieg. Der Übergangsstruktur (ÜS) für die Umwandlung cis→trans entspräche in diesem Fall dem lokalen Maximum zwischen den beiden Minima.
Mindestens genauso interessant ist jedoch die Suche nach Übergangsstrukturen, also nach lokalen Maxima (bzw. Sattelpunkten in mehreren Dimensionen). Die Energiedifferenz zwischen stabilen Minimumsstrukturen und Übergangsstrukturen (Aktivierungsenergie, ) bestimmt maßgeblich die Geschwindigkeit chemischer Reaktionen und Umwandlungen.
Funktionsweise von Photoschaltern¶
Wie eingangs erwähnt, bezeichnet man als Photoschalter ein Molekül, das durch Licht zwischen zwei definierten Zuständen
umgeschaltet werden kann.
Diese beiden Zustände unterscheiden sich in ihrer räumlichen Struktur – und damit auch in ihren chemischen bzw.
physikalischen Eigenschaften.
Das Umschalten erfolgt durch Licht, da die Barrieren zwischen den beiden Zuständen im Grundzustand so hoch sind, dass
sie durch die thermische Energie bei Raumtemperatur nicht überwunden werden können.
Die grundlegende Funktionsweise eines Photoschalters lässt sich wie folgt zusammenfassen:
Das Molekül befindet sich zunächst in einem Ausgangszustand (z. B. der trans-Form bei Azobenzol).
Trifft Licht der passenden Energie auf das Molekül, wird es absorbiert – das Molekül gelangt in einen angeregten elektronischen Zustand.
In diesem angeregten Zustand kann sich die räumliche Struktur ändern (z. B. Rotation an einer Doppelbindung). Dadurch „schaltet“ das Molekül in den zweiten Zustand um.
Nach der strukturellen Veränderung entspannt das Molekül wieder in einen neuen Grundzustand (z. B. cis-Form).
Da cis- und trans-Form unterschiedliche Eigenschaften besitzen (etwa Farbe, Polarität oder Reaktivität), lässt sich das Molekül wie ein Schalter verwenden – mit Licht als Signal zum Ein- bzw. Umschalten.
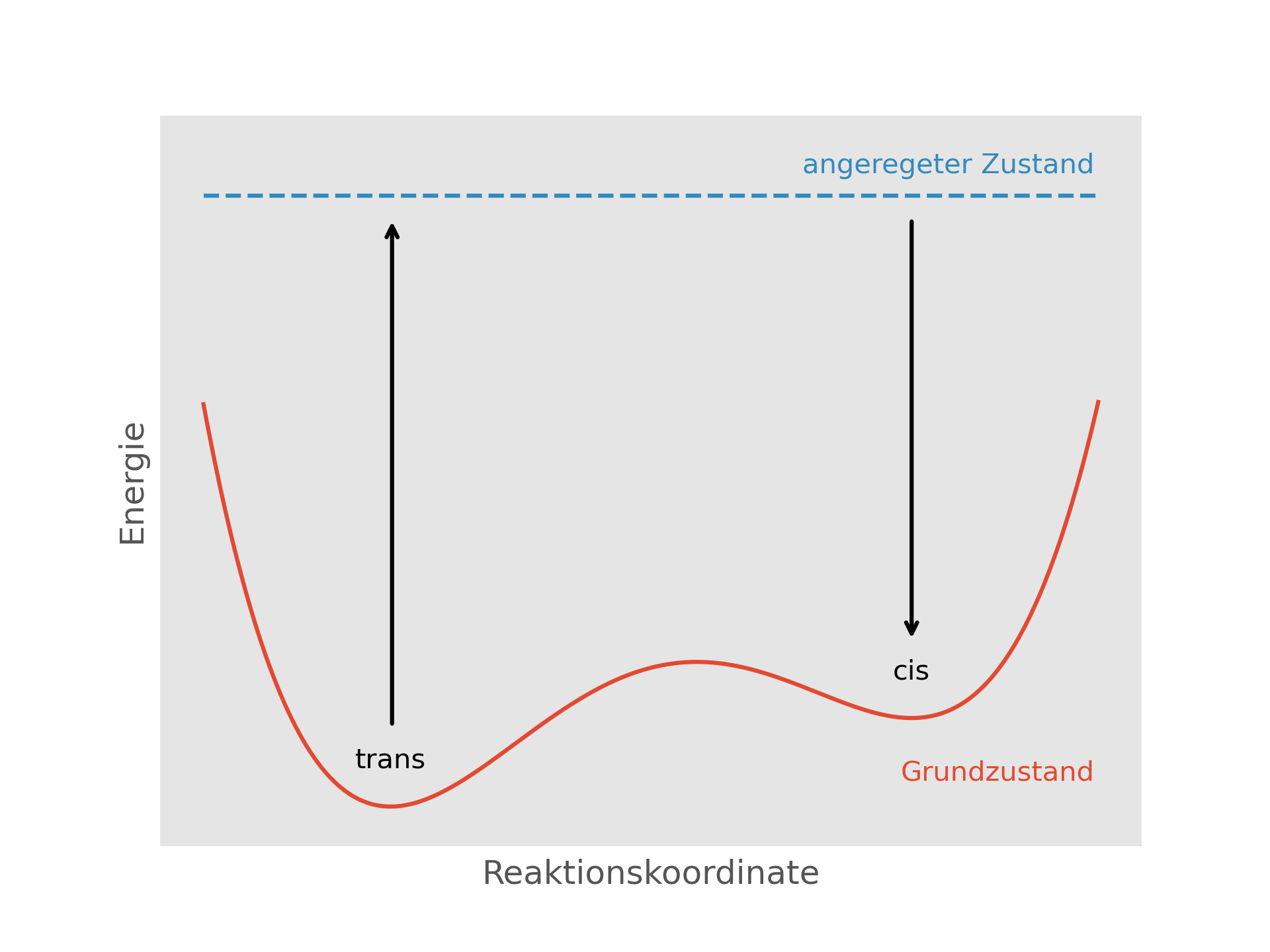
Schematische Darstellung der Funktionsweise eines Photoschalters. Der Wechsel zwischen cis- und trans-Form erfolgt nicht im Grundzustand durch Überwinden des Übergangszustandes. Stattdessen wird das Molekül zunächst in einen angeregten elektronischen Zustand angeregt, in dem die Umwandlung stattfinden kann.